






















ANSYS BLOG
February 20, 2019
Overview of the Regulatory Pathway for In Silico Testing of Medical Devices
Medical device manufacturers have traditionally relied on bench testing, animal testing and clinical trials (i.e., human testing) to establish the safety and efficacy of medical devices.
This is true both internally, when making decisions about various aspects of a device design, and externally when applying for regulatory approval.
These tests can be expensive and time-consuming. Additionally, the ability of animal testing to predict the safety and efficacy of human treatments has come into question.
The introduction of regulatory
pathways for in silico evaluation
will make simulation an increasing
trend in the healthcare industry
The use of computational modeling (i.e., simulation or in silico trials) is being used more often in the design of medical devices. These simulations accelerate innovation and provide comprehensive evidence of long-term safety. However, in spite of the benefits of simulation, the utilization of computational modeling as evidence in regulatory submissions is lagging.
With the introduction of regulatory pathways for in silico testing of medical devices, we can expect to see simulation as an increasing trend in the regulatory side of the healthcare industry.
Not Long Ago, In Silico Medical Device Testing Was Futuristic
I encountered the discrepancy between the utilization of modeling and regulatory inclusion in healthcare as a biomedical engineer at Fluent, Inc. (now part of Ansys).
It was 2001 and I was straight out of graduate school. During this time, I was supporting computational fluid dynamics (CFD) users at various medical device industry accounts, including the Office of Science and Engineering Labs (OSEL) at the U.S. Food and Drug Administration’s (FDA) Center for Devices and Radiological Health (CDRH).
Using in silico testing for
regulatory submissions was
generally viewed as futuristic
It was very clear from the start that modeling was generally viewed more as a research tool and less as a credible source of information that could be a component of regulatory submissions.
In fact, to consider modeling as a tool that was on equal footing with other forms of device evaluation was futuristic — even futurist — thinking.
The Benefits of In Silico Testing of Medical Devices
Fast forward to 2011, when the CDRH establishes computational modeling a regulatory science priority. This was formal recognition of the ability of in silico testing to provide a much deeper understanding of device performance.
In fact, in silico studies have some distinct and appealing advantages over the other forms of device evaluation. These advantages include being able to conduct parametric, statistical and patient-specific analyses of a device’s performance in a highly controlled manner.

In silico testing will be able to
test edge cases, so the health of
all patients will be considered
when evaluating the device
Even edge cases can be created and explored via simulation, providing some understanding of the ability of a device to operate under the most extreme situations. This is quite the opposite of animal testing or human trials, where it can be difficult to obtain statistically significant results at the far edges of the physiological envelope.
Over several years, the simulation community has made significant progress in the regulatory science of computer modeling. Critical barriers related to the utilization of modeling with regulatory agencies, like the FDA, have also been addressed. These barriers were related to model validation and model reporting.
FDA Explains How to Write Computational Modeling and Simulation Reports
The first hurdle to establishing the regulatory application of simulation was the lack of a well-established structure to summarize a computational model’s framework and results for review.
To address this, the FDA published guidance that describes the format and contents of a computational modeling and simulation (CM&S) report.
A primary goal of this guidance is to ensure that a sponsor includes all the information required to judge whether a model has sufficient credibility to serve as valid scientific evidence for regulatory decision-making.
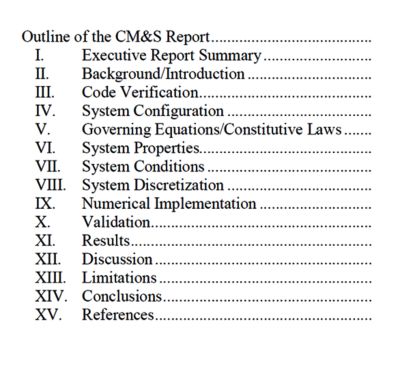
The CM&S report outline — as
provided by the FDA Guidance
The document also provides a common language for the device industry and regulators to use when planning, discussing and reviewing the various elements of a computational modeling study.
The main body of the guidance describes the general format of the report which can be applied to most physics disciplines. To aid the user, the guidance also includes a series of appendices that provide detailed reporting recommendations for CFD, finite element analysis (FEA), electromagnetics (EM), ultrasound, heat transfer and optics.
For example, the appendix on computational electromagnetics recommends that the user provide a summary of the governing equations utilized (e.g., Maxwell’s equations) and information about the electrical properties of the device and tissues being modeled.
Assessing the Risk of In Silico Medical Device Testing
Another reason for the limited utilization of simulation results in regulatory submissions was the need for consensus on the evidentiary bar required to establish sufficient credibility of a computational model. The American Society of Mechanical Engineers (ASME) verification and validation (V&V) subcommittee on computational models of medical devices (ASME V&V 40 subcommittee) was formed in 2011 to address this critical need.
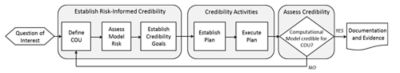
The risk-informed credibility assessment framework,
as defined by the ASME V&V 40 standard
Comprised of members from the medical device industry, industry service providers, software developers and the FDA, the group developed the risk-informed credibility assessment framework.
The main tenet of the framework is that high-risk decisions based on a computer model require more validation than lower-risk decisions.
Therefore, the ASME standard guides how the user determines the amount of V&V that is required to establish model credibility. This standard complements the ASME V&V standards for FEA, CFD and heat transfer.
How to Validate In Silico Medical Device Testing
After seven years of hard work, the ASME standard on the V&V of medical devices was published Nov. 19, 2018. This standard completes the regulatory pathway for computational modeling.
The ASME V&V 40 subcommittee is now working to educate the community about the standard and its best practices. This education is being done through industry days, training sessions, examples and new work items.
As a vice chair of the ASME V&V 40 subcommittee for medical devices, it has been my honor and great pleasure to work with such a dedicated group of talented scientists and engineers.

The ASME V&V 40 subcommittee
wants to teach in silico testing
best practices so that industry can
reliably simulate medical devices
for internal decision-making
and regulatory submission
For more on how Ansys can help simulate medical applications, read In Silico Testing or the industry page on Healthcare: Medical Device Simulation.
To learn how to join the ASME V&V 40 subcommittee, visit its contact page.